作者:Yang, W.; Li, P.; Duanmu, K.; Zhao, Z.; Tian, L.; Zhai, D.-D.; Sherman, B. D.; Humphrey, M. G.; Shi, Z.-J.*; Zhang, C.*; Hu, K.*
期刊: Angew. Chem. Int. Ed. 2026, e9951729
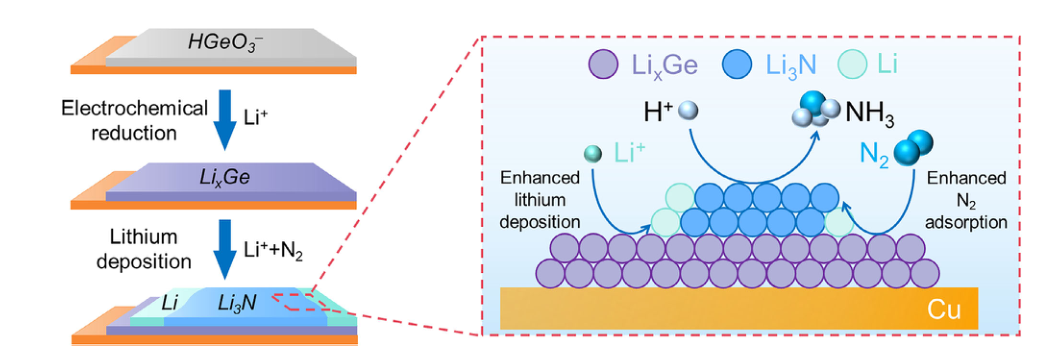

作者:Yang, W.; Li, P.; Duanmu, K.; Zhao, Z.; Tian, L.; Zhai, D.-D.; Sherman, B. D.; Humphrey, M. G.; Shi, Z.-J.*; Zhang, C.*; Hu, K.*
期刊: Angew. Chem. Int. Ed. 2026, e9951729
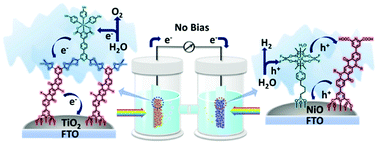
作者:Tian, L.; Liu, R.; Zhao, Z.; Li, P.; Yang, W.; Niu, F.; Hu, K.*
期刊: Chinese Chemical Letters 2026, 112521
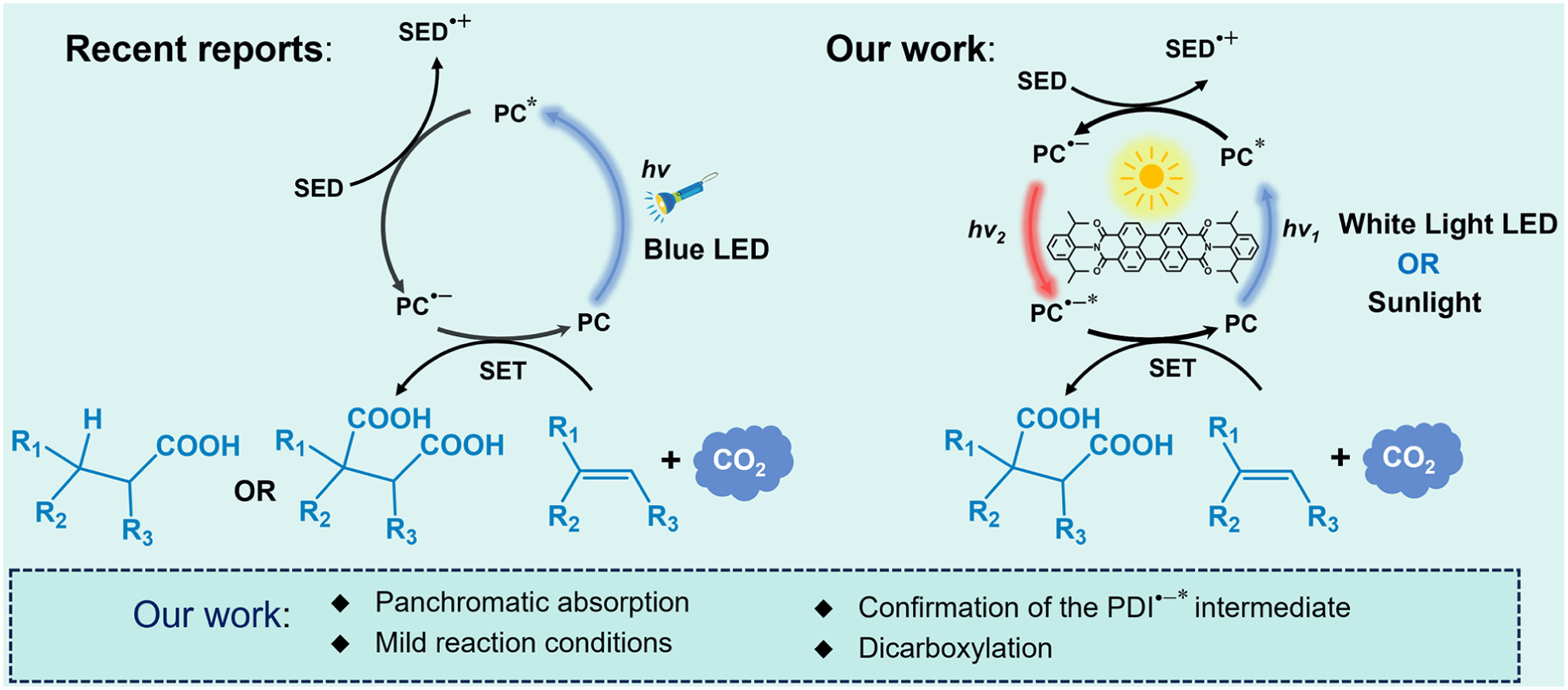
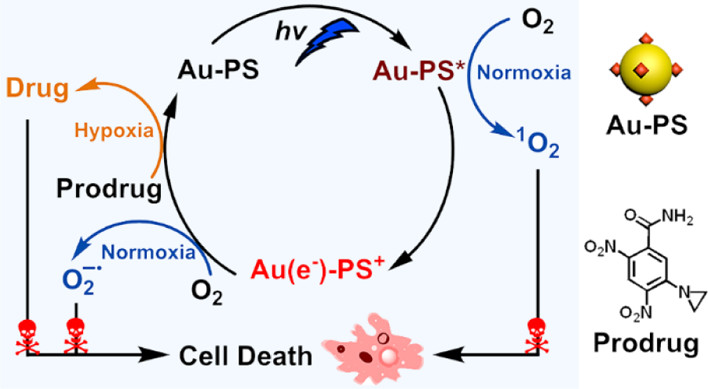
作者: Li, P.; Zhao, Z.; Tian, L.; Liu, R.; Wang, X.; Yang, W.; Cui, Y.-S.; Humphrey, M. G.; Zhang, C.*; Hu, K.*
期刊:J. Am. Chem. Soc. 2025, 147, 44817-44824
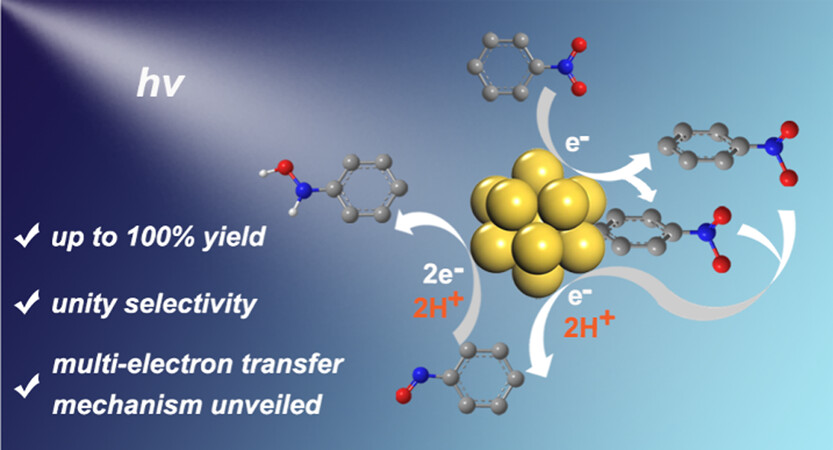
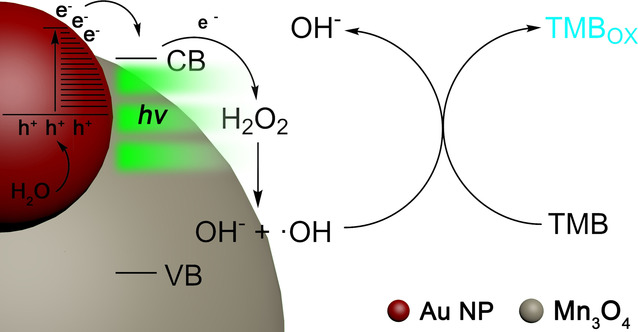
作者:Yang, W.; Li, P.; Han, Y.; Zhao, Z.; Tian, L.; Zhang, Z.; Humphrey, M. G.; Zhang, C.*; Hu, K.*
期刊: Chem. Sci. 2025, 16, 20229-20238

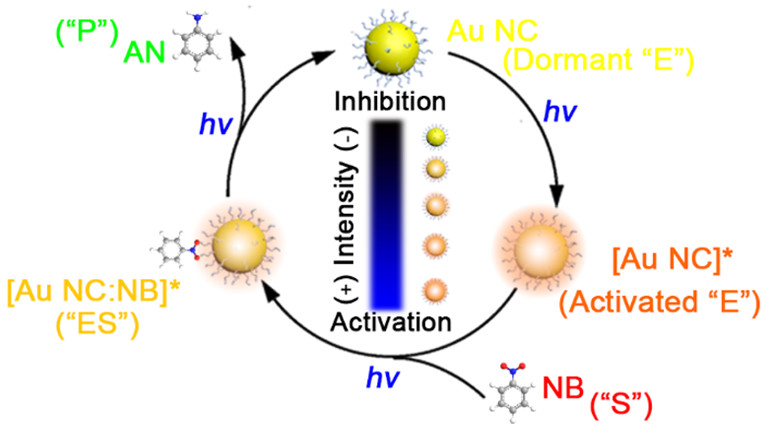
作者:Yang, W.; Li, P.; Zhao, Z.; Tian, L.; Niu, F.; Sampaio, R. N.*; Hu, K.*
期刊:CHEM-ASIAN J. 2025, 20, e00768
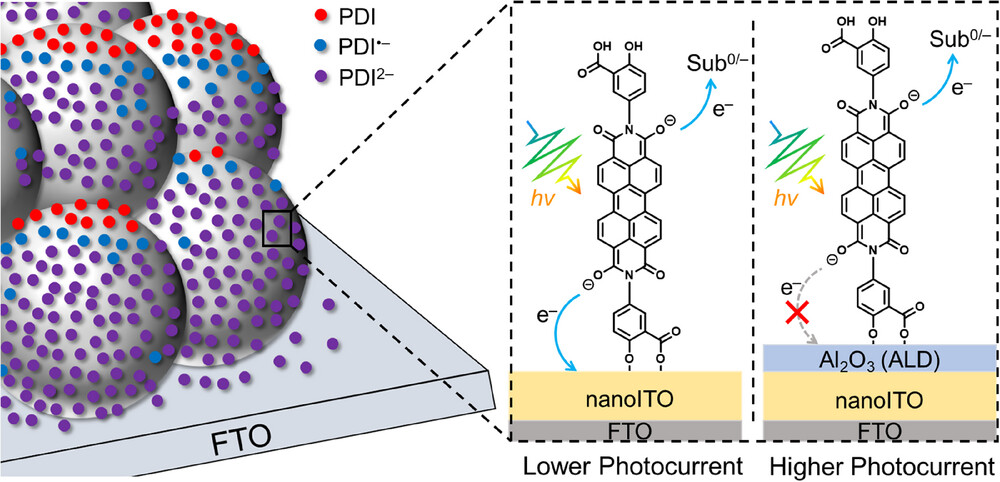
作者:Li, P.; Bourgois, C.; Glaser, F.; De Kreijger, S.; Cadranel, A.; Troian-Gautier, L.*; Hu, K.*
期刊:J. Am. Chem. Soc. 2025, 147, 12082-12091
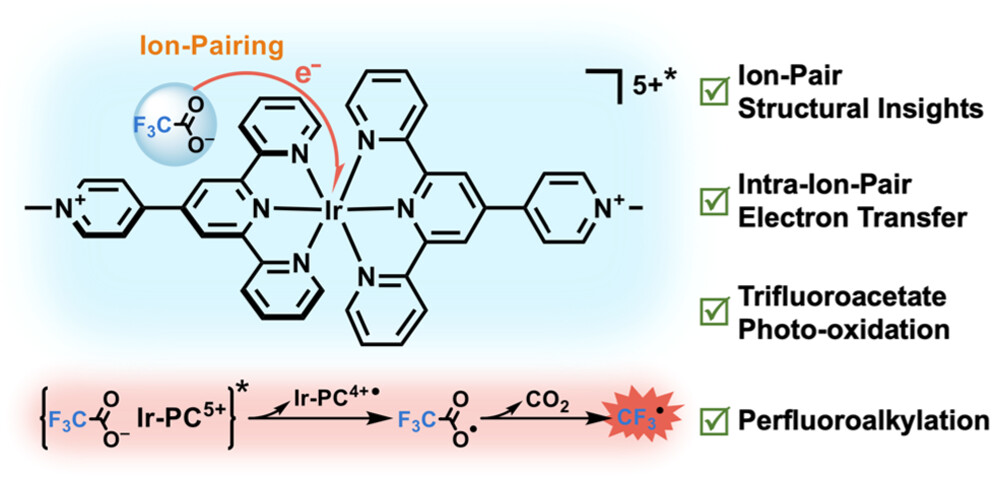
作者:Zhao, Z.; Li, J.; Yuan, W.; Cheng, D.; Ma, S.; Li, Y.; Shi, Z; Hu, K.*
期刊:J. Am. Chem. Soc, 2023, 146 (2), 1364-1373 (Front Cover)

作者: Li, P.; Liu, R.; Zhao, Z.; Yang, W.; Niu, F.; Tian, L.; Hu, K.*
期刊:ACS Sustainable Chem. Eng. 2024, 12, 1, 10–17
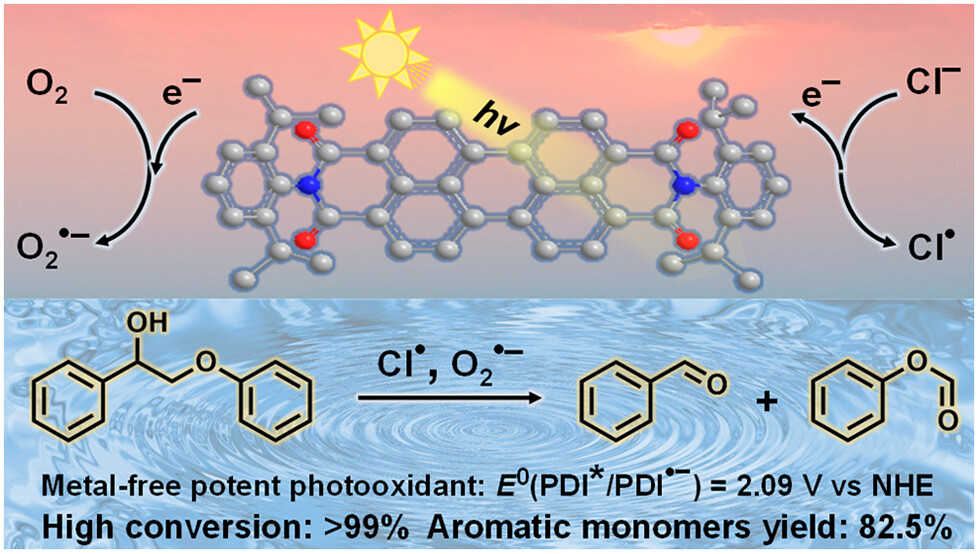
作者: Kang, W.; Li, B.; Zhao, Z.; Sun, S.; Feng, C.; Hu, K.*; Houk, K.N.; Guo, H.*
期刊:ACS Catalysis 2023 13 (20), 13588-13596.

作者: Weng, W.; Lin, Z.; Zhang, H.; Niu, F.; Wang, C.; Hu, K*; Guo, J.*
期刊:JACS Au 2023, 3, 12, 3391–3399.
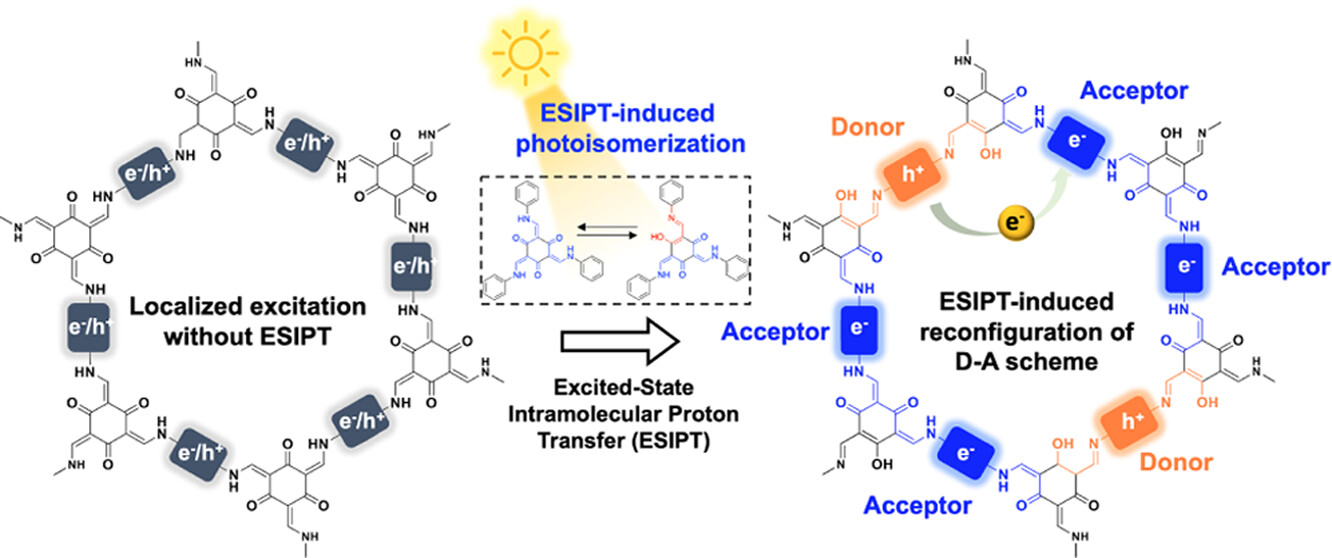
作者: Sun, X.; Liu, j.; Wang, H.; Li, Q.; Zhou, J.; Li, P.; Hu, K*; Wang, C*.;Jiang, B.*
期刊:Chem. Eng. J., 2023, 472, 144750
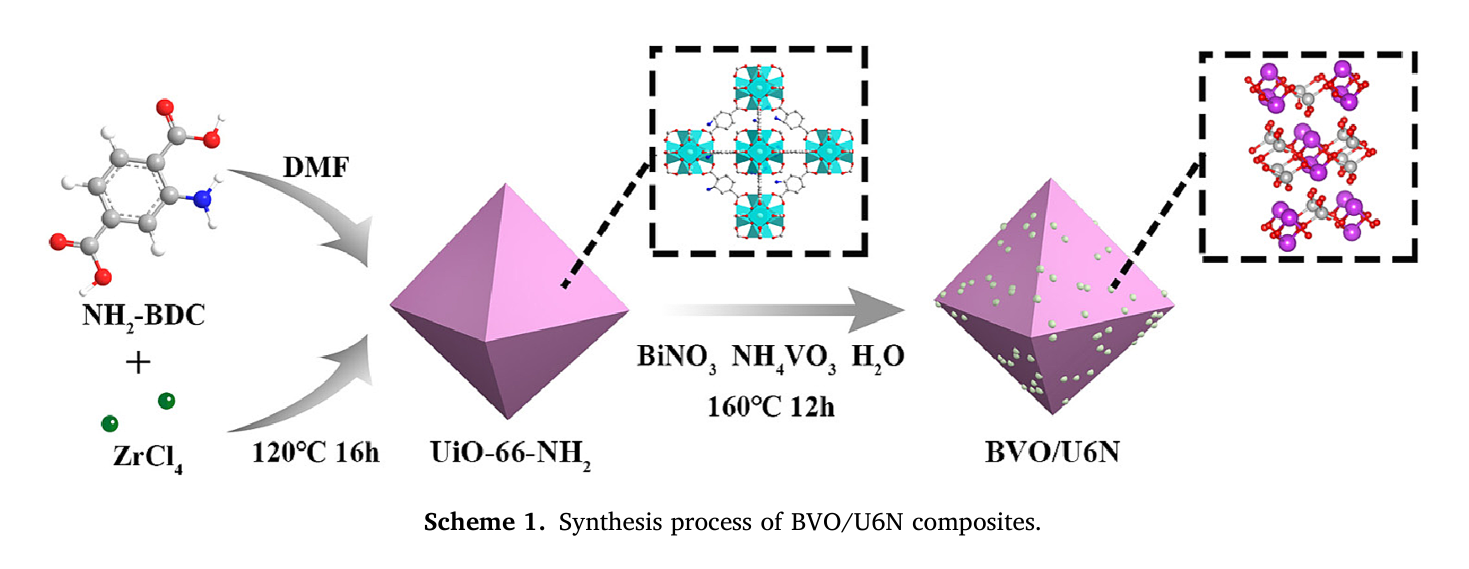
作者: Han, Y.; Chang, M.; Zhao, Z.; Niu, F.; Zhang, Z.; Sun, Z.; Zhang, L.; Hu, K.*
期刊:ACS Appl. Mater. Interfaces 2023, 15, 9, 11678–11690.
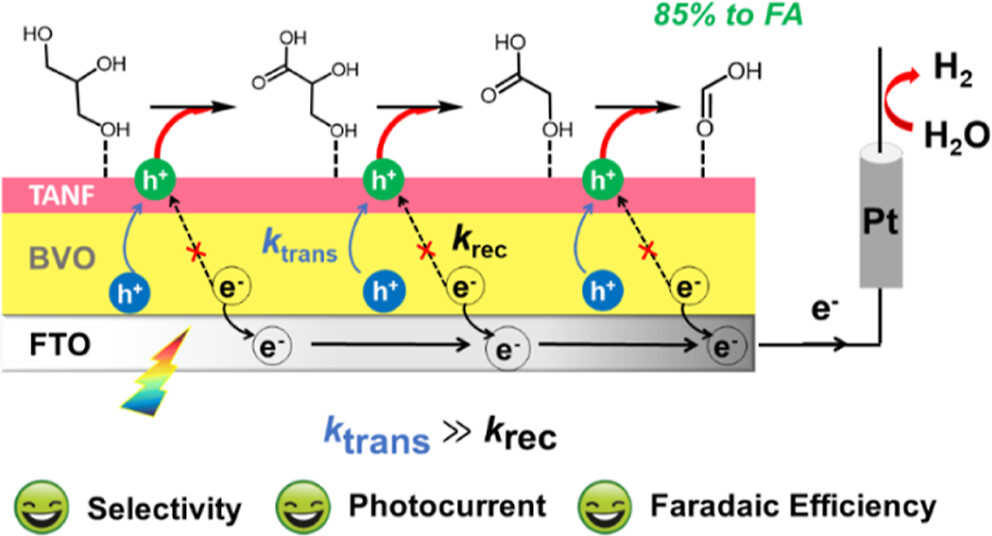
作者: Niu, F.; Zhang, P.; Zhang, Z.; Zhou, Q.; Li, P.; Liu, R.; Li, W.; Hu, K.*
期刊:J. Mater. Chem. A, 2023, 11, 4170.
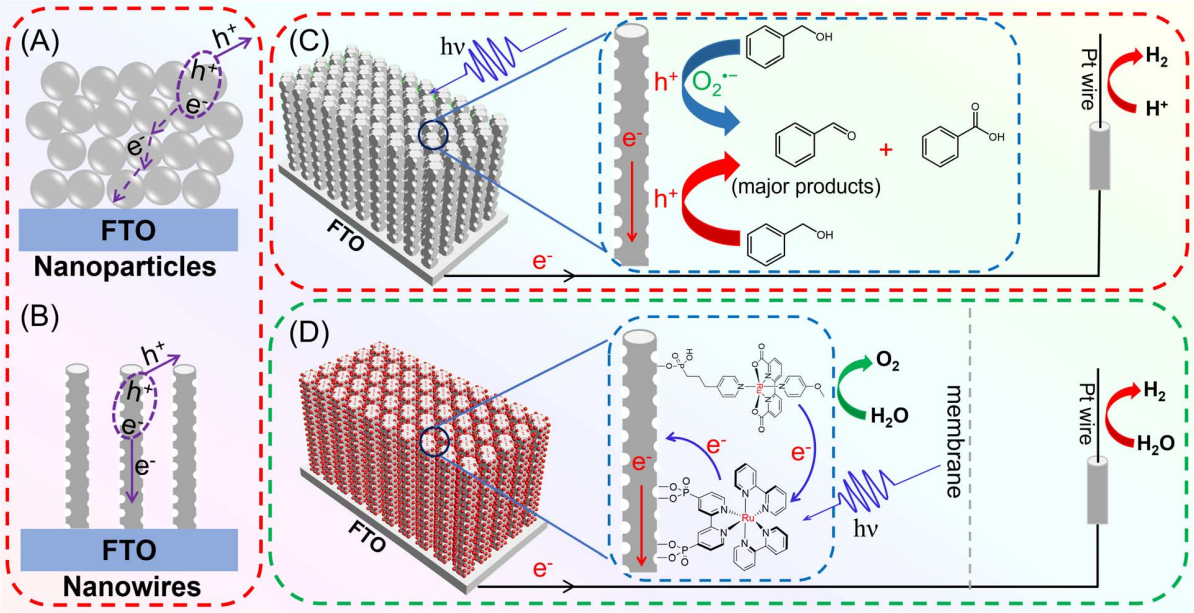
作者: Li, P.; Liu, R.; Zhao, Z.; Niu, F.; Hu, K.*
期刊:Chem. Commun., 2023, 59, 1777
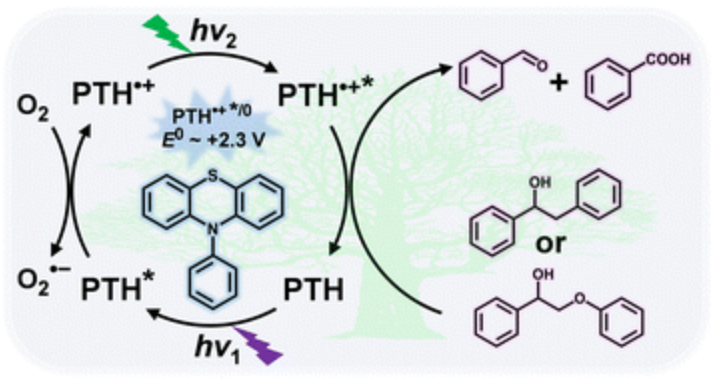
作者: Wang, X.; Liu, R.; Tian, L.; Bao, J.; Zhao, C.; Niu, F.; Cheng, D.; Lu, Z.; Hu, J.; Meyer, G. J.*; Hu, K.*
期刊:J. Phys. Chem. C 2022, 126, 18374−18382.
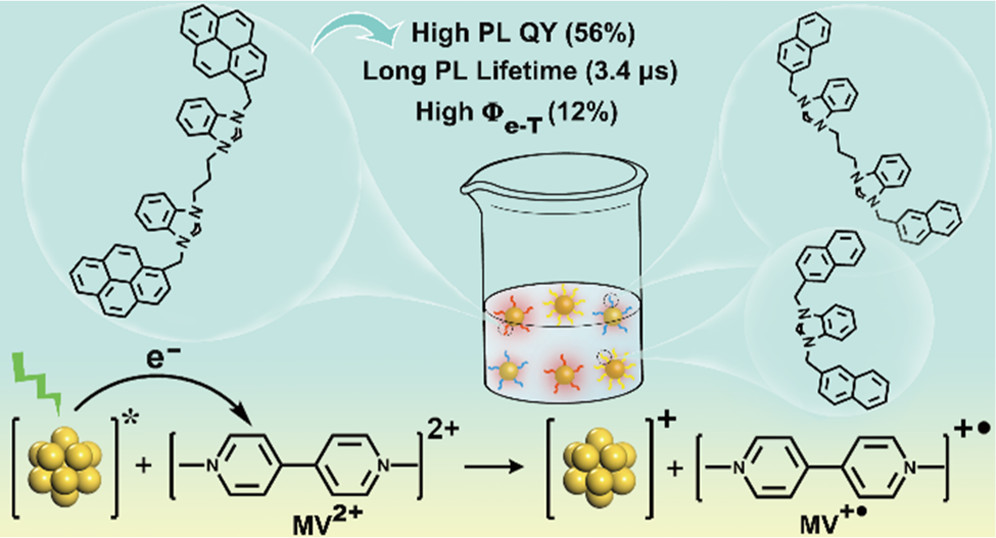
作者: Li, P.; Deetz, A. M.; Hu, J.; Meyer, G. J.*; Hu, K.*
期刊:J. Am. Chem. Soc. 2022, 144, 17604-17610.
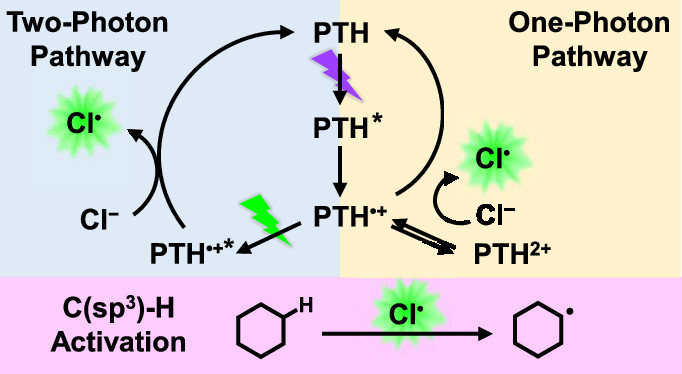
作者: Niu, F.; Zhou, Q.; Han, Y.; Liu, R.; Zhao, Z.; Zhang, Z.; Hu, K.*
期刊:Acs Catal. 2022, 12,16, 10028-10038.
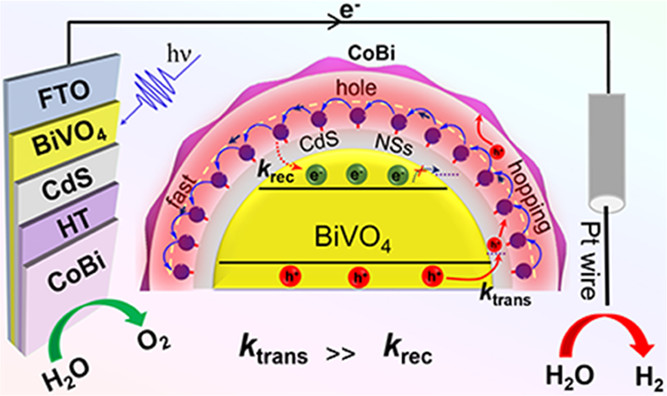
作者: Zhao, Z.; Niu, F.; Li, P.; Wang, H.; Zhang, Z.; Meyer, G. J.*; Hu, K.*
期刊:J. Am. Chem. Soc. 2022, 144, 7043-7047 (Front Cover).

作者: Song, Y.; Li, A.; Li, P.; He, L.; Xu, D.; Wu, F.; Zhai, F.; Wu, Y.; Hu, K.*; Wang, S.*; Sheridan, M. V.*
期刊:Chem. Mater. 2022, 34, 2771-2778.
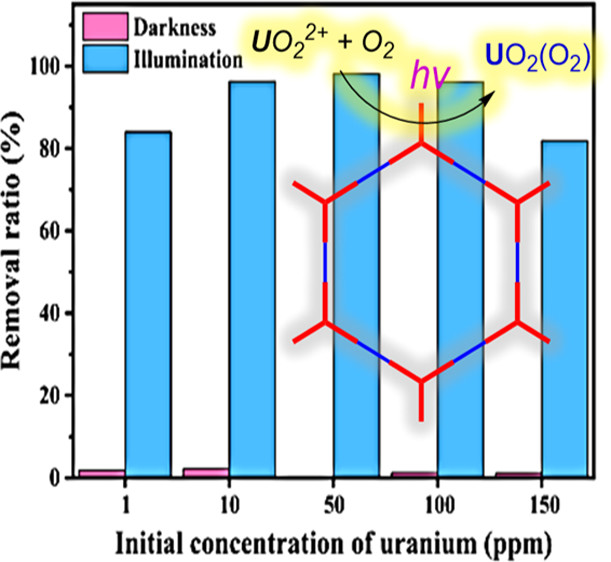
作者: Niu, F.; Zhou, Q.; Liu, R.; Hu, K.*
期刊:ACS Appl. Energy Mater. 2022, 5 (1), 1244-1251.
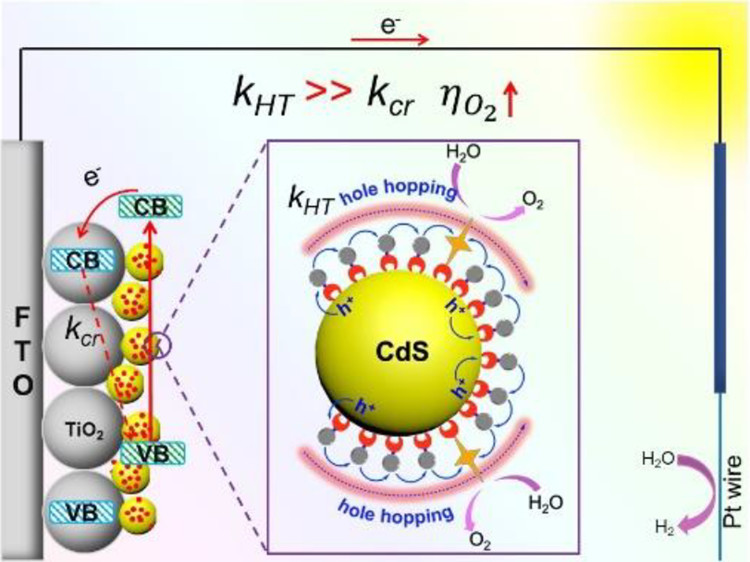
作者: Cheng, D.; Liu, R.; Tian, L.; Zhou, Q.; Niu, F.; Yue, Y.*; Hu, K.*
期刊:ACS Appl. Nano Mater. 2021, 4 (12), 13413-13424.
作者:Shen, L.; Zhang, S.; Ding, H.; Niu, F.; Chu, Y.; Wu, W.*; Hu, Y.; Hu, K.*; Hua, J.* Hu, K.*; Hua, J.*
期刊:Chem. Commun. 2021, 57, 5634-5637.
作者: Han, X.; Liu, R.; Zhang, H.; Zhou, Q.; Feng, W.*; Hu, K.*
期刊:Chem. Asian J. 2021, 16, 1603-1607.
作者: Liu, R.; Cheng, D.; Zhou, Q.; Niu, F.; Hu, K.*
期刊:ACS Appl. Nano Mater. 2021, 4, 990-994.
作者: Hu, K.; Sampaio, R. N.; Schneider, J.; Troian-Gautier, L.; Meyer, G. J.*
期刊:J. Am. Chem. Soc. 2020, 142 (38), 16099-16116.
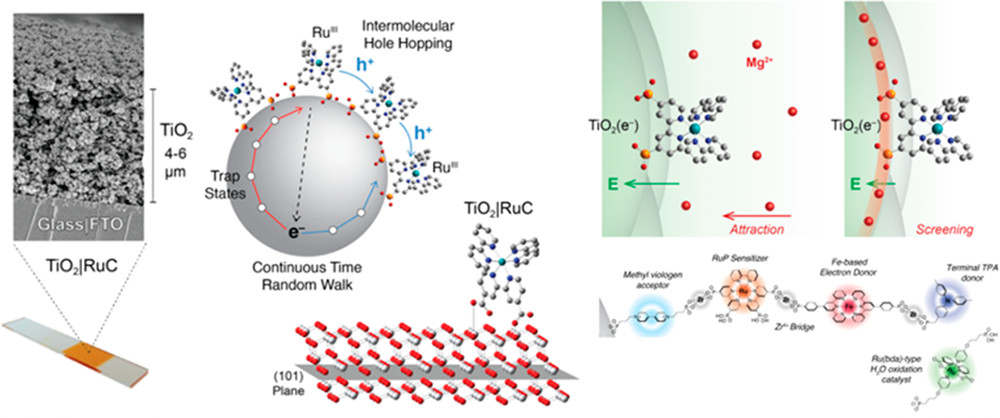
作者: Shi, Y.; Wang, R.; Yuan, W.; Liu, Q.; Shi, M.; Feng, W.; Wu, Z.; Hu, K.*; Li, F.*
期刊:ACS Mater. Lett. 2019, 1, 418-424.
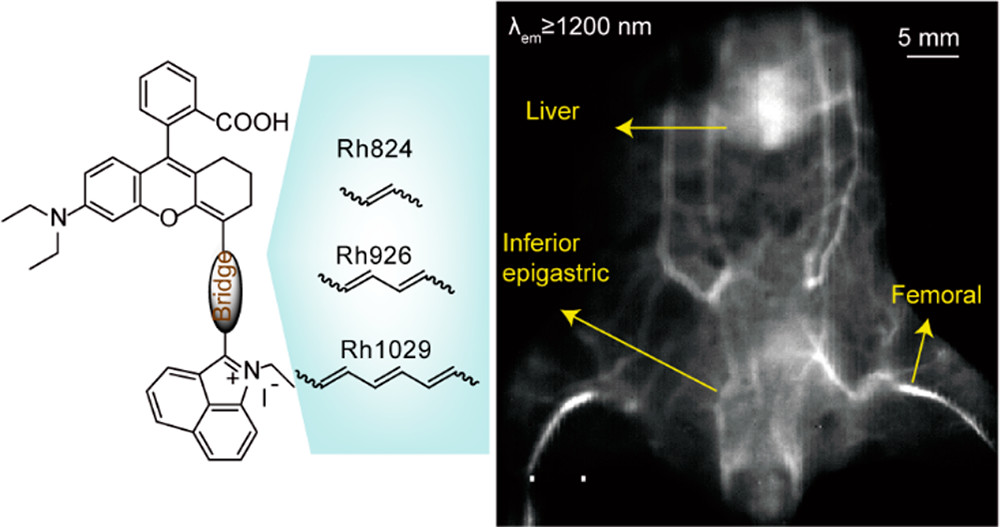
作者: Shi, Y.; Wang, R.; Yuan, W.; Liu, Q.; Shi, M.; Feng, W.; Wu, Z.; Hu, K.*; Li, F.*
期刊:ACS Appl. Mater. Interfaces 2018, 10, 20377-20386
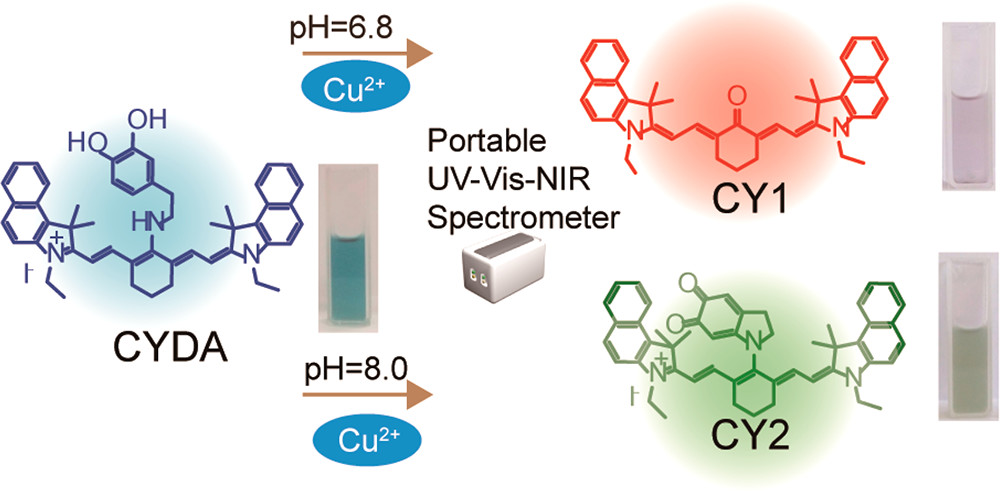
作者: Hu, K.*; Sheiko, S. S.*
期刊:Chem. Commun. 2018, 54, 5899-5902.
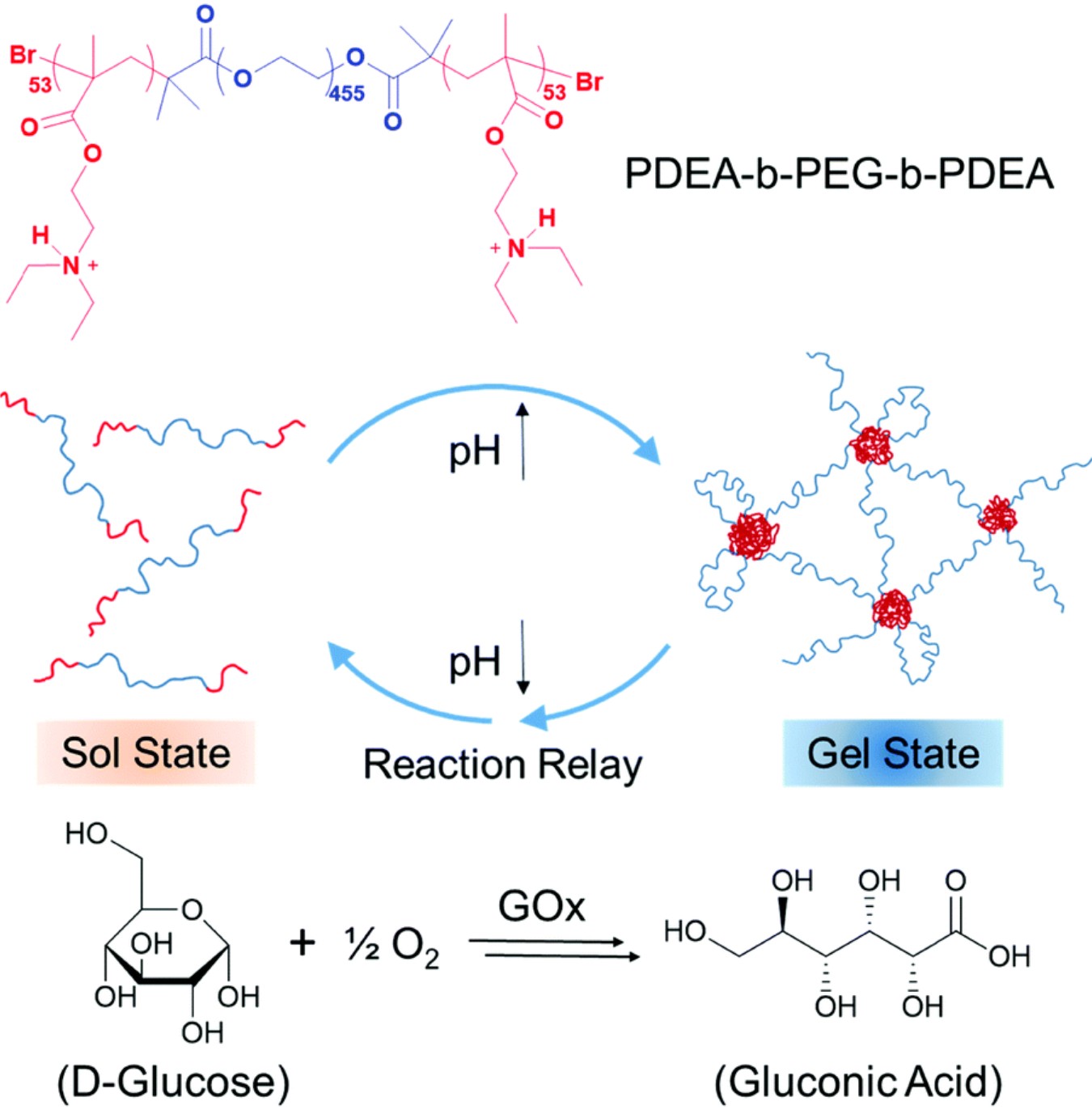
作者: Hu, K. #; Sampaio R. N. #; Marquard S. L.; Tamaki Y.; Meyer T. J.; Meyer G. J.
期刊:Inorg. Chem. 2018, 57, 486-494.
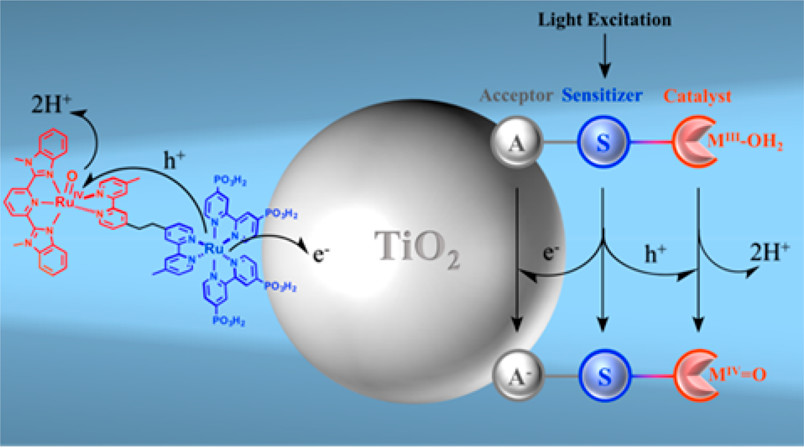
作者: Nayak, A.#; Hu, K.#; Roy, S; Brennaman, M. K.; Shan, B.; Gish, M.; Meyer, G. J.; Meyer, T. J.
期刊:J. Phys. Chem. C 2018, 122, 13455-13461
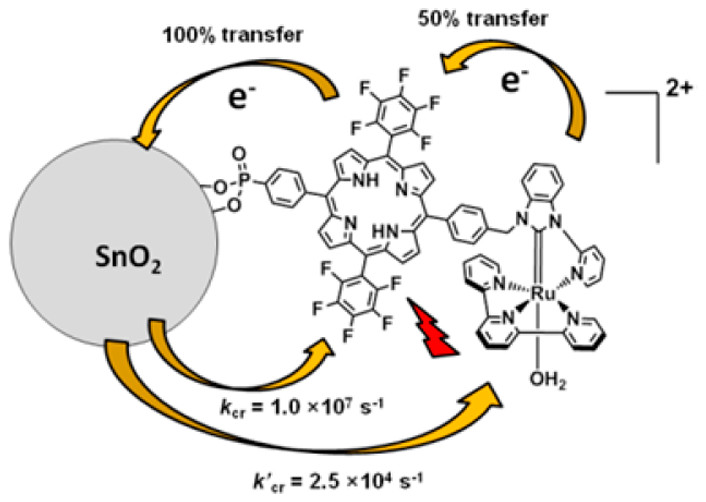
作者: Hu, K.; Blair, A. D.; Piechota, E. J.; Schauer, P. A.; Sampaio, R. N.; Parlane, F. G. L.; Meyer, G. J.; Berlinguette, C. P.
期刊:Nature Chem. 2016, 8, 853-859.
作者: Hu, K.; Meyer, G. J.
期刊:Langmuir 2015, 31, 11164.
作者: Hu, K.; Robson, K. C. D.; Beauvilliers, E. E.; Schott, E.; Zarate, X.; Arratia-Perez, R.; Berlinguette, C. P.; Meyer, G. J.
期刊:J. Am. Chem. Soc. 2014, 136, 1034.
作者: Hu, K.; Robson, K. C. D.; Johansson, P. G.; Berlinguette, C. P.; Meyer, G. J.
期刊:J. Phys. Chem. C 2014, 118, 17079.
作者: Hu, K.; Robson, K. C. D.; Berlinguette, C. P.; Meyer, G. J.
期刊:Thin Solid Films 2014, 560, 49.
作者: Hu, K.; Robson, K. C. D.; Johansson, P. G.; Berlinguette, C. P.; Meyer, G. J.
期刊:J. Am. Chem. Soc. 2012, 134, 8352.
作者: Ye, H.; Wang, Z.; Hu, K.; Wu, W.; Gong, X.; Hua, J.*
期刊:Science China Chemistry 2022, 65 (1), 170-181.
作者: Pan, C.; Chao, J.; Niu, F.; Xie, S.; Gu, H.; Su, T.; Hu, K.; Zhang, D.-W.*; Liu, K.; Liu, G.; Xie, T.; Li, Z.-T.*; Zhang, L.*
期刊:Advanced Materials Interfaces 2022, 9 (1), 2101678.
作者: Mi, Z.; Zhou, T.; Weng, W.; Unruangsri, J.; Hu, K.; Yang, W.; Wang, C.; Zhang, K. A. I.; Guo, J.*
期刊:Angew. Chem. Int. Ed. 2021, 60, 9642-9649
作者: Wang, D.; Eberhart, M. S.; Sheridan, M. V.; Hu, K.; Sherman, B. D.; Nayak, A.; Wang, Y.; Marquard, S. L.; Dares, C. J.; Meyer, T. J.
期刊:Proc. Nat. Acad. Sci. USA 2018, DOI: 10.1073/pnas.1802903115
作者: Sampaio, R. N.; Piechota, E. J.; Troian-Gautier, L.; Maurer, A. B.; Hu, K.; Schauer, P. A.; Blair, A. D.; Berlinguette, C. P.; Meyer, G. J.
期刊:Proc. Nat. Acad. Sci. USA 2018, 115, 7248-7253.
作者: Piechota, E. J.; Troian-Gautier, L.; Sampaio, R. N.; Brennaman, M. K.; Hu, K.; Berlinguette, C. P.*; Meyer, G. J.*
期刊:J. Am. Chem. Soc. 2018, 140 (23), 7176-7186.
作者: Maurer, A. B.; Hu, K.; Meyer, G. J.
期刊:J. Am. Chem. Soc. 2017, 139, 8066
作者: Swords, W. B.; Simon, S. J. C.; Parlane, F. G. L; Dean, R. K.; Kellett, C. W; Hu K.; Meyer, G. J.; Berlinguette, C. P.
期刊:Angew. Chem. Int. Ed. 2016, 55, 1.
作者: Simon, S. J. C.; Parlane, F. G. L.; Swords, W. B.; Kellett, C. W.; Du, C.; Lam, B.; Dean, R. K.; Hu, K.; Meyer, G. J.; Berlinguette, C. P.
期刊:J. Am. Chem. Soc. 2016, 138, 10406.
作者: DiMarco, B. N.; Motley, T. C.; Balok, R. S.; Li, G.; Siegler, M. A.; O’Donnell, R. M.; Hu, K.; Meyer, G. J.
期刊:J. Phys. Chem. C 2016, 120, 14226.
作者: Li, G.; Hu, K.; Robson, K. C. D.; Gorelsky, S. I.; Meyer, G. J.; Berlinguette, C. P.; Shatruk, M.
期刊:Chem. Eur. J. 2015, 21, 2173.
作者: Cottingham, P.; Wallace, D. C.; Hu, K.; Meyer, G. J.; McQueen T. M.
期刊:Chem. Commun. 2015, 51, 7309.
作者: Robson, K. C. D.; Hu, K.; Meyer, G. J.; Berlinguette, C. P.
期刊:J. Am. Chem. Soc. 2013, 135, 1961.
作者: Li, G.; Hu, K.; Yi, C.; Knappenberger, Jr., K. L.; Meyer, G. J.; Gorelsky, S. I.; Shatruk, M.
期刊:J. Phys. Chem. C 2013, 117, 17399.
作者: Cui, Y. H.; Xue, M. Z.; Wang, X. L.; Hu, K.; Fu, Z. W.
期刊:Electrochem. Commun. 2009, 11, 1045.